
CHÚ THÍCH: Một số từ viết tắt:
NOD- Bệnh tiểu đường nonobese;
IDDM- Tiểu đường phụ thuộc insulin;
IL- Interleukin;
TNF- Yếu tố hoại tử khối u;
TCR- Thụ thể tế bào T;
TdT- Đầu cuối dideoxynucleotidetransferase;
TUNEL- Nick X-dUTP qua trung gian TdT và nhãn kết thúc;
T1D- Tiểu đường type I;
β – Beta, tế bào b.
Bệnh tiểu đường type 1 (loại 1)
Bệnh tiểu đường nói chung thường chia thành các loại sau:
- Tiểu đường type 1 (T1D) (do phá hủy tế bào beta tụy, dẫn đến thiếu insulin tuyệt đối).
- Tiểu đường type 2 (do giảm chức năng của tế bào beta tụy tiến triển trên nền tảng đề kháng insulin).
- Tiểu đường thai kỳ (là tiểu đường được chẩn đoán trong 3 tháng giữa hoặc 3 tháng cuối của thai kỳ và không có bằng chứng về tiểu đường type 1, tiểu đường type 2 trước đó).
- Tiểu đường do các nguyên nhân khác, như: Tiểu đường sơ sinh hoặc tiểu đường do sử dụng thuốc và hóa chất như sử dụng glucocorticoid, điều trị HIV/AIDS hoặc sau cấy ghép mô...[1].
Bệnh tiểu đường loại 1 do thiếu hụt insulin gây ra do mất các tế bào b (beta-β) tuyến tụy sản xuất insulin, thường phát triển ở giới trẻ, và chiếm khoảng 5% –10% dân số mắc tiểu đường trên toàn thế giới. Sự phát triển của bệnh tiểu đường loại 1 là hậu quả của sự phá hủy tế bào b tiến triển bởi quá trình tự miễn dịch, thời kỳ không có triệu chứng thường kéo dài trong nhiều năm [2].
Cơ chế gây bệnh tiểu đường type 1
Tiểu đường type 1 là một thể bệnh nặng. Nguyên nhân là do tế bào beta của tiểu đảo Langerhans bị tổn thương gây nên tình trạng thiếu insulin tuyệt đối (hình 1).
- Cơ chế qua trung gian miễn dịch: Quá trình tổn thương tế bào bêta là quá trình tự miễn dịch. Những cá nhân có tính mẫn cảm di truyền sẽ tăng nguy cơ bị tiểu đường type 1 sau một tấn công của môi trường bên ngoài như virus quai bị, sởi, coxsakie B4 và B5, retro loại C...
- Những cá thể có mang kháng nguyên HLA B8, B15 nhất là DR3, DR4, DR3/DR4 sẽ tăng nguy cơ bị tiểu đường type 1.
Các yếu tố môi trường trên sẽ tấn công những cá thể có tố bẩm di truyền đối với tiểu đường type 1. Chỉ một tổn thương rất nhỏ của tế bào bêta cũng làm giải phóng ra kháng nguyên, kích thích cơ thể sinh tự kháng thể gây hoạt hóa phản ứng viêm tiểu đảo tự miễn. Các kháng nguyên có thể là GAD (glutamic acid decarboxylase) một protein Kd nằm trong bào tương của tế bào bêta. Tự kháng thể sẽ phản ứng với kháng nguyên.
Đại thực bào lympho được hoạt hoá sẽ tập trung quanh tiểu đảo của tụy gây ra phản ứng viêm. Tế bào lympho T tiết ra các hoá chất trung gian trong đó có interleukin-1 gây ảnh hưởng độc với tế bào bêta. Interleukin-1 cảm ứng hình thành các gốc tự do làm tế bào bêta bị tổn thương và phá hủy dẫn đến ngừng tiết insulin [1].
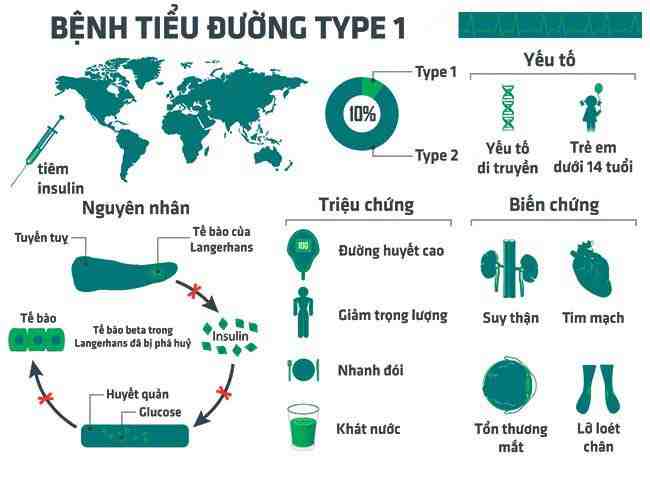
Hình 1. Sơ đồ mô phỏng của bệnh tiểu đường type 1.
Cơ chế phá hủy tế bào beta đảo tụy
Một công trình nghiên cứu trên chuột thí nghiệm cho thấy: Tế bào T CD8 + và CD4 + tự hoạt động đã được chỉ định những vai trò quan trọng độc lập trong việc tiêu diệt các tế bào beta sản xuất insulin dẫn đến bệnh tiểu đường loại 1. Mặc dù CD4 giúp tạo ra các phản ứng hiệu quả của tế bào T CD8 + trong mô bạch huyết đã được mô tả rộng rãi, nhưng liệu hai quần thể tế bào này có hợp tác với nhau trong việc phá hủy đảo tại chỗ hay không vẫn chưa rõ ràng. Bằng cách sử dụng kính hiển vi 2-photon trong một mô hình chuột mắc bệnh tiểu đường, chúng tôi đã hình dung được cả quần thể tế bào T tác động trong tuyến tụy khi bệnh khởi phát. Tế bào trợ giúp CD4 + T cho thấy khả năng bị bắt giữ trong mô ngoại tiết cao hơn nhiều so với CD8 + dành riêng cho tiểu đảoTế bào T. Sự bắt giữ gia tăng này là do phức hợp tương thích mô chính (MHC) phụ thuộc vào lớp II và tương quan cục bộ với việc tuyển chọn tế bào trình diện kháng nguyên. Tế bào T CD8 + bị mất CD4 tiếp tục trợ giúp đặc biệt trong tuyến tụy, thông qua việc ngăn chặn sự nhận dạng MHC lớp II, không thể duy trì các chức năng hiệu ứng tối ưu, góp phần cản trở sự tiến triển của bệnh tiểu đường. Do đó, chúng tôi cung cấp thông tin chi tiết mới về cơ chế tế bào điều chỉnh chức năng của tế bào T hiệu ứng trong các mô ngoại vi, có ý nghĩa quan trọng đối với liệu pháp miễn dịch.
Sự phát triển của bệnh T1D phản ánh sự thất bại của cơ chế dung nạp miễn dịch kiểm soát các tế bào T tự phản ứng. Mặc dù thành phần chính xác của tế bào miễn dịch trong tổn thương tiểu đường thay đổi theo thời gian, tế bào T thường chiếm ưu thế ở bệnh nhân T1D. Cả CD4 + trợ giúp và CD8 + tế bào T gây độc tế bào (CTL) đều có mặt, và có một lượng lớn bằng chứng chỉ ra rằng cả hai đều là tác nhân chính trong sự phát triển của T1D. Thật vậy, hầu hết các locus nhạy cảm đã được xác định đều chứa các gen ứng cử viên liên quan đến sự hoạt hóa và điều hòa tế bào T, và phức hợp tương thích mô chính cụ thể (MHC) lớp II và ở mức độ thấp hơn, các haplotype MHC lớp I có khuynh hướng phát triển bệnh tiểu đường.
Bệnh khởi phát liên quan đến hai bước tuần tự trong các mô khác biệt về mặt giải phẫu. Đầu tiên, các tế bào T tự hoạt động, đã trốn tránh các điểm kiểm tra dung nạp, được kích hoạt trong các hạch bạch huyết dẫn lưu tuyến tụy (pLN) bởi các tế bào trình diện kháng nguyên (APC) hiển thị các kháng nguyên tế bào beta. Ở giai đoạn này, CD4 + tế bào T được yêu cầu để kích hoạt hiệu quả CD8 + tế bào T. Thứ hai, các tế bào T tác động được kích hoạt xâm nhập vào tuyến tụy. Trong khi các tế bào T CD4 + của tác động xâm nhập được cho là góp phần làm chết tế bào beta thông qua hoạt hóa đại thực bào, tế bào T CD8 + có thể tiêu diệt trực tiếp tế bào beta theo cách phụ thuộc vào kháng nguyên. Ngoài ra, cả hai tế bào T effector tham gia vào các tiếp xúc phụ thuộc kháng nguyên với APC, được yêu cầu để duy trì chức năng của effector theo thời gian trong tuyến tụy, ít nhất là đối với tế bào T CD8 + . Những tương tác này dường như rất quan trọng đối với việc duy trì các đặc tính gây độc tế bào ở ngoại vi.
Động lực hoạt hóa tế bào T trong các cơ quan lympho thứ cấp, bao gồm pLN, đã được nghiên cứu rộng rãi trong hơn một thập kỷ trên các mô hình chuột sử dụng kính hiển vi hai photon. Các nghiên cứu này đã cung cấp một khuôn khổ để hiểu được vai trò của các dạng tương tác động khác nhau với APC, dẫn đến cảm ứng dung nạp hoặc mồi tế bào T. Hơn nữa, họ nhấn mạnh tầm quan trọng của sự hợp tác giữa các tế bào T trong việc tạo ra các phản ứng miễn dịch. Đặc biệt, các nghiên cứu gần đây đã tiết lộ một vũ đạo phức tạp của các tương tác tuần tự giữa các APC, CD4 + và CD8 +Tế bào T trong các cơ quan bạch huyết trong quá trình mồi và sự tồn tại của một loại phụ tế bào đuôi gai (DC) chuyên biệt có thể đóng vai trò là nền tảng cho sự hợp tác tế bào T CD4 + và CD8 + . Động lực học tế bào T CD8 + hoặc CD4 + gây tiểu đường đã được nghiên cứu ở đảo tụy. Tuy nhiên, việc hình dung các phản ứng của tế bào T trong các cơ quan sâu như tuyến tụy in vivo vẫn còn nhiều thách thức và sự hợp tác chức năng giữa các quần thể tế bào T CD8 + hoặc CD4 + trong tuyến tụy trong thời gian T1D vẫn chưa được khám phá.
Do đó, trong nghiên cứu này, chúng tôi bắt đầu tìm hiểu cách các tế bào T CD8 + và CD4 + của cơ quan tác động hợp tác trong việc phá hủy đảo nhỏ trong giai đoạn khởi phát T1D. Để cho phép điều này, các phương pháp tiếp cận hình ảnh trong ổ được áp dụng trực tiếp vào tuyến tụy trên mô hình chuột mắc bệnh tiểu đường tự miễn dịch, trong đó cả tế bào T CD8 + và CD4 + đều được yêu cầu để gây bệnh.
Dòng chảy năng động của các loại tế bào miễn dịch khác nhau trong đảo tụy dẫn đến phá hủy tế bào beta. Trong khi các tế bào lympho T CD8 + và CD4 + thường đại diện cho quần thể tế bào miễn dịch ưu thế và độc lập đóng vai trò quan trọng trong việc tiêu diệt tế bào beta, liệu chúng có thể cộng tác trong quá trình phá hủy đảo nhỏ hay không vẫn chưa được biết. Bằng cách sử dụng kính hiển vi hai photon in vivo để hình dung các tế bào T CD8 + và CD4 + đặc hiệu HA trong tuyến tụy của chuột biểu hiện HA trong tế bào beta, chúng tôi nhận thấy rằng các tế bào T CD4 + hiển thị sự bắt giữ tăng phụ thuộc MHC lớp II trong mô ngoại tiết. CD4+ Bắt giữ tế bào T trong nước tương quan với APC tuyển dụng, mà xâm nhập CD4 + tế bào T là đủ để kích hoạt. Cuối cùng, các tế bào T CD8 + của tác nhân gây thâm nhiễm không được tiếp tục trợ giúp thông qua điều trị bằng kháng thể kháng MHC lớp II không duy trì được các chức năng tối ưu của cơ quan tác động đặc biệt trong tuyến tụy, cùng với việc giảm sự thâm nhập của tế bào T tác động, cản trở sự tiến triển của bệnh tiểu đường. Những kết quả này hỗ trợ một vai trò quan trọng đối với các tế bào trợ giúp CD4 + T của effector trong việc duy trì các chức năng effector của CTL tại các vị trí đích.
Theo các nghiên cứu trước đây về viêm cách điện không đồng nhất. Đáng chú ý, các đảo nhỏ bị xâm nhập bởi một vài tế bào T chứa cả tế bào T CD8 + và CD4 + , cho thấy rằng thời gian xâm nhập là tương tự đối với cả hai loại tế bào T, mặc dù tỷ lệ khác nhau. Cả hai quần thể tế bào T đều di chuyển bên ngoài các đảo nhỏ với vận tốc trung bình tương tự như vận tốc được mô tả đối với tế bào T ở các mô ngoại vi. Khả năng di chuyển của chúng tăng lên theo mức độ xâm nhập, và sự bắt giữ ở các đảo nhỏ cao, phù hợp với sự nhận biết kháng nguyên. Ngược lại với tế bào T CD8 + , CD4 +Tế bào T cho thấy sự bắt giữ rõ rệt trong mô ngoại tiết, tương quan với việc tuyển dụng cả DC và đại thực bào. Việc tuyển dụng các APC tại địa phương có thể phục vụ các mục đích khác nhau. Đầu tiên, các đại thực bào được tuyển dụng có thể tham gia trực tiếp vào quá trình tiêu diệt tế bào beta. Thứ hai, các APC được tuyển chọn có thể thực bào các tế bào đã giết chết, thúc đẩy sự trình bày kháng nguyên tế bào beta và khuếch đại sự hoạt hóa tế bào T. Cuối cùng, sự tập hợp cục bộ của các APC có thể hạn chế sự di chuyển của tế bào T để tạo điều kiện cho việc cảm ứng hoặc duy trì các chức năng của cơ quan tác động và tăng sinh CTL.
Đáp ứng lâu dài của tế bào T CD8 + phụ thuộc vào sự trợ giúp của CD4 trong các mô không phải lympho. Bằng chứng cho thấy các DC là những người hòa giải chính trong quá trình này. Đầu tiên, hầu hết các tế bào biểu hiện MHC lớp II trong mô ngoại tiết ở đây tương ứng với CD103 + DCs. Thứ hai, cả tế bào T CD8 + và CD4 + đặc hiệu với kháng nguyên tế bào beta đã được chứng minh là độc lập tham gia vào các tương tác qua trung gian kháng nguyên với DC. Cuối cùng, điều trị chống MHCII làm giảm sự bắt giữ tế bào T. Trong khi việc phong tỏa nhận dạng kháng nguyên trực tiếp làm suy giảm tương tác tế bào T / APC CD4 + , làm giảm CD8 +Sự bắt giữ tế bào T gợi ý một hiệu ứng gián tiếp. Kể từ khi CD4 + / APC cụm có thể thu hút CD8 + tế bào T, CD8 + tế bào T có thể có nhiều khả năng APC xúc khi họ đang hoặc đã được tiếp xúc với CD4 + tế bào T, như mô tả trong LN. Điều này có thể giải thích tại sao các cụm CD4 + / DC / CD8 + có thể được phát hiện thường xuyên bằng các thí nghiệm đồng tiêu. Tuy nhiên, các thử nghiệm sâu hơn sẽ được yêu cầu để đánh giá tầm quan trọng của loại tương tác ba tế bào trong việc phân phối trợ giúp.
Cuối cùng, điều trị kháng thể kháng MHCII tại thời điểm khi sự hoạt hóa do kháng nguyên điều khiển của các tế bào T đặc hiệu với HA trong pLN đã xảy ra và các tế bào T tác dụng đã thâm nhập vào tuyến tụy ảnh hưởng đến sự biểu hiện của các dấu hiệu liên kết với các chức năng của tác nhân hiệu ứng trong CD8 đặc hiệu với HA + Tế bào T. Điều này hỗ trợ vai trò quan trọng đối với sự trợ giúp liên tục của tế bào T CD4 + , qua trung gian của APC, trong việc duy trì hiệu quả tiêu diệt tế bào T CD8 + trong tuyến tụy. Các CTL được tạo với sự trợ giúp của CD4 trong pLN do đó có thể không được lập trình để giữ lại các chức năng của bộ phận tạo hiệu ứng trong tuyến tụy và cần được trợ giúp liên tục để có chức năng tối ưu. Phù hợp với sự thoát mạch của tế bào T CD4 + phụ thuộc kháng nguyên trong tuyến tụy được tạo điều kiện thuận lợi bởi DCs, điều trị chống MHCII làm suy giảm khả năng tuyển dụng tế bào T CD4 + . Điều thú vị là số lượng tế bào T CD8 + cũng bị ảnh hưởng, phản ánh vai trò của tế bào T CD4 + trong việc tuyển dụng và / hoặc tăng sinh tế bào T CD8 + trong tuyến tụy. Do đó, sự chậm trễ trong tăng đường huyết và sự ổn định sau đó của cả mức đường huyết và sức khỏe chung, do đó có thể phản ánh tác động kết hợp của việc giảm thâm nhập tế bào tác động và giảm khả năng duy trì sự phá hủy tế bào beta của tế bào T CD8 + .
Các cơ chế chính xác giúp phân phối cơ bản khi nhận biết kháng nguyên trong tuyến tụy vẫn cần được nghiên cứu thêm. Trong LNS, CD40 trung gian cấp phép của DCs bởi CD4 + T tế bào gây ra sự biểu hiện của các phân tử đồng kích thích và tạo thành con đường chính của CD4 giúp đỡ cho ngây thơ CD8 + mồi tế bào. Tuy nhiên, các yêu cầu đối với kích thích tế bào T CD8 + của effector có thể ít nghiêm ngặt hơn. Hỗ trợ điều này, các DC tuyến tụy từ những con chuột được điều trị bằng thuốc chống MHCII hiển thị mức độ tương tự của các phân tử đồng kích thích như đối chứng (các quan sát chưa được công bố của chúng tôi). Ngoài ra, sự thật rằng CTL kháng u dựa vào IL-2 và IFNγ để đào thải khối u và HNT CD4 + của tuyến tụy. Tế bào T ở đây có thể tiết ra IFNγ và IL-2 để đáp ứng với kháng nguyên cho thấy rằng những cytokine này có thể tham gia vào việc giúp nhân bản 4 tế bào T CD8 + . Một triển vọng trêu ngươi là các DC có thể đóng vai trò như một nền tảng để cung cấp các cytokine này.
Tóm lại, chúng tôi tiết lộ vai trò quan trọng đối với sự trợ giúp tiếp tục của CD4 trong việc duy trì chức năng của tế bào T CD8 + trong tuyến tụy và trong tiến triển bệnh tiểu đường tự miễn và do đó tiết lộ vai trò chưa từng có đối với sự hợp tác giữa tế bào T CD4 + và CD8 + có thể có ý nghĩa quan trọng để thiết kế các chiến lược điều trị mới chống lại T1D [3].
Một bài báo khác viết về “Quá trình chết rụng tế bào β trong bệnh tiểu đường tự miễn qua trung gian tế bào T”, xin được dịch tóm tắt như sau:
Chúng ta biết rằng IDDM là kết quả của sự phá hủy có chọn lọc các tế bào β tuyến tụy, sản xuất insulin. Sự phá hủy này được điều phối bởi các tế bào T CD4 + đặc hiệu với kháng nguyên tế bào β sản xuất ra các cytokine tiền viêm. Các tế bào β bị giết chết như thế nào vẫn chưa được hiểu rõ. Điều này là do sự chết của tế bào β rất khó phát hiện trong cơ thể sống, do động học chậm của quá trình viêm và quá trình loại bỏ nhanh chóng các tế bào chết khỏi vật chủ. Thâm nhiễm tế bào lympho, được gọi là viêm cách điện, diễn ra trong vài tháng; các tổn thương ban đầu chủ yếu là viêm cách điện nhẹ, ít mất tế bào β. Vào thời điểm chuột NOD có biểu hiện tăng đường huyết kéo dài, > 90% tổng khối lượng tế bào β bị phá hủy, khiến việc phân tích không thể thực hiện được. Do đó, rất khó để thiết lập một cửa sổ có thể tái tạo để nghiên cứu quá trình chết tế bào β in vivo .
Một số nhóm đã tìm cách nghiên cứu sự chết của tế bào β trong ống nghiệm, chẳng hạn như thử nghiệm xem một số cytokine tiền viêm có thể giết chết tế bào β hay không. Chưa đạt được sự đồng thuận về cách tế bào β chết, nhưng interleukin (IL) -1β, một mình hoặc kết hợp với interferon-γ và yếu tố hoại tử khối u (TNF) -α, có thể ức chế sự bài tiết insulin do glucose gây ra và gây ra sự sản sinh độc tố, oxit nitric, bởi tế bào β. Điều này dẫn đến suy đoán rằng việc sản xuất IL-1β tại chỗ có thể đóng một vai trò quan trọng trong việc gây chết tế bào β. Trong khi một số nghiên cứu đã xem xét vai trò của các cytokine — đặc biệt là IL-2, IL-10, IL-12, TNF-α và interferon-γ — để tăng cường bệnh lý tiểu đảo và tăng tốc độ chết của tế bào β in vivo, họ đã không đưa ra bất kỳ đánh giá trực tiếp nào về việc tế bào β bị ảnh hưởng như thế nào và liệu chúng có chết trên thực tế hay không. Hơn nữa, rất khó để đánh giá mức độ phản ánh của các mô hình cytokine này đối với quá trình bệnh tự nhiên [4].
Nghiên cứu về “Bộ nhớ hoạt động tự động / Bộ nhớ Tế bào CD4 + và CD8 + xâm nhập vào các đảo ghép và nội sinh ở chuột NOD bị tiểu đường có cách sử dụng tế bào T tương tự” cho thấy như sau: Sự hiểu biết sâu sắc về bộ phận tiếp nhận tế bào T (TCR) của các tế bào T tác động thúc đẩy quá trình tự miễn dịch tái phát sẽ hỗ trợ sự phát triển của liệu pháp miễn dịch để ngăn ngừa thải ghép tiểu đảo. Theo đó, chúng tôi đã sử dụng chiến lược đo tế bào dòng đa tham số để đánh giá chuỗi biến số TCR β (Vβ) của các tập con tế bào T liên quan đến quá trình thải ghép tiểu đảo qua trung gian tự miễn dịch ở chuột nhận NOD bị tiểu đường. Naïve CD4 + và CD8 +Tế bào T thể hiện một danh mục TCR đa dạng, giống nhau ở tất cả các mô được kiểm tra ở những người nhận NOD bao gồm tuyến tụy và mô ghép đảo nhỏ. Mặt khác, hiệu ứng / bộ nhớ CD8 + tế bào T trong ghép đảo bị chi phối bởi một đến bốn chuỗi TCR Vβ, và việc sử dụng chuỗi TCR Vβ cụ thể khác nhau giữa người nhận. Tương tự, tế bào T CD4 + T tế bào T CD4 + của bộ nhớ ghép xâm nhập đảo nhỏ biểu hiện một số lượng hạn chế các chuỗi TCR Vβ phổ biến, mặc dù nhìn chung, tính đa dạng tiết mục TCR đã tăng lên so với tế bào T CD8 + của bộ nhớ / bộ nhớ . Đáng chú ý, phần lớn những người nhận NOD cho thấy sự gia tăng hiệu ứng mang TCR Vβ12 / bộ nhớ CD4 +Tế bào T trong mảnh ghép đảo nhỏ, hầu hết trong số đó đã tăng sinh, cho thấy sự mở rộng vô tính. Điều quan trọng là, việc sử dụng TCR Vβ bởi tác động / bộ nhớ tế bào T CD4 + và CD8 + thâm nhập vào mô ghép đảo cho thấy sự giống nhau nhiều hơn so với tiết mục được tìm thấy trong tuyến tụy trái ngược với việc dẫn lưu hạch thận, hạch tụy hoặc lá lách. Các kết quả này cùng nhau chứng minh rằng các tế bào T CD4 + và CD8 + làm trung gian cho quá trình thải ghép tự miễn dịch tự miễn dịch của bộ nhớ được đặc trưng bởi việc sử dụng chuỗi TCR Vβ bị hạn chế, và tương tự như các tế bào T thúc đẩy sự phá hủy các đảo nhỏ nội sinh.
Ở người và chuột NOD, một mô hình tự phát cho T1D, tự miễn dịch tế bào β được xem như là một phản ứng viêm mạn tính trung gian bởi autoreactive CD4 + và CD8 + tế bào T. Khởi đầu phản ứng gây tiểu đường liên quan đến việc tế bào T nhận ra một số lượng hạn chế các tự kháng nguyên tế bào β. Khi quá trình tự miễn dịch của tế bào β tiến triển, một số tự kháng nguyên được nhắm mục tiêu do sự lan truyền biểu mô trong và giữa các phân tử, dẫn đến sự mở rộng nhiều kiểu dòng của tế bào T tác động đặc hiệu tế bào β gây bệnh (Teff). Loại thứ hai được thể hiện rõ ràng bởi một tiết mục thụ thể tế bào T (TCR) được đánh dấu bằng sự biểu hiện của nhiều gen biến TCR (V) bởi các tế bào T cư trú trên đảo, và các dòng tế bào T đặc hiệu tế bào β. Một khi ∼80% khối lượng tế bào β đã bị phá hủy và / hoặc không hoạt động, nồng độ đường huyết tăng cao sẽ đạt được và sự khởi đầu của bệnh tiểu đường được chẩn đoán công khai [5].
Tìm hiểu chất dẫn truyền tín hiệu và chất kích hoạt phiên mã 1 (Signal transducer and activator of transcription 1, được viết tắt là: STAT-1) đóng một vai trò quan trọng trong việc phá hủy tế bào beta do cytokine gây ra như thế nào, một nghiên cứu đã thực hiện phân tích proteome của các đảo nhỏ chuột C57Bl / 6 và STAT-1 (- / -) tiếp xúc với cytokine và các protein được ưu tiên cho tiềm năng của chúng liên quan đến T1D. Kết quả: Nhiều protein điều chỉnh STAT-1 đã được xác định và chia thành các nhóm khác nhau theo chức năng sinh học của chúng. Nhóm protein lớn nhất là nhóm tham gia vào quá trình tổng hợp và xử lý protein. Phân tích mạng cho thấy sự tương tác phức tạp giữa các protein từ các nhóm chức năng khác nhau và phân tích IPA đã xác nhận tác dụng bảo vệ của việc xóa STAT-1 đối với sự chết tế bào beta do cytokine gây ra. Cuối cùng, vai trò trung tâm trong cơ chế điều chỉnh STAT-1 này được giao cho chất điều chỉnh nhỏ liên quan đến ubiquitin 4 (SUMO4). Những phát hiện này xác nhận vai trò trung tâm của STAT-1 trong sự phá hủy gây ra viêm đảo tụy và quan trọng nhất là làm sáng tỏ các con đường protein cơ bản liên quan [6].
Một công trình nghiên cứu khác cho thấy: Phân tích các phần tuyến tụy được thu hoạch từ những cá nhân có T1D đã cho thấy khả năng thâm nhập miễn dịch hoàn toàn trong các đảo nhỏ riêng lẻ, chứng minh vai trò quan trọng của tế bào T CD4 và CD8 trong việc phá hủy tế bào beta. Điều này trái ngược hẳn với các phần tuyến tụy của những người mắc bệnh T2D, mặc dù có mức độ cao của dấu hiệu viêm toàn thân, nhưng không có sự thâm nhập tế bào T tương tự trong các đảo nhỏ của tuyến tụy. Hầu như tất cả những người phát triển T1D trước 5 tuổi đều sản sinh ra tự kháng thể đặc hiệu với insulin (IAA), cho thấy vai trò quan trọng của các peptit có nguồn gốc từ phân tử insulin trong cơ chế bệnh sinh. Các tự kháng thể đảo là một dấu hiệu chẩn đoán phân biệt giữa T1D và T2D và phát sinh từ tương tác tế bào B tự hoạt động và tế bào T CD4 tự hoạt động. Các alen loại II của kháng nguyên bạch cầu (HLAs) ở người DR4, DQ8 và DQ2 gây nguy cơ di truyền cao nhất cho bệnh T1D ở bệnh nhân. Sự liên kết mạnh mẽ giữa alen HLA II với T1D cho thấy rằng các tế bào T CD4 hạn chế HLA II đóng một vai trò quan trọng trong cơ chế bệnh sinh. Tế bào T CD4 có thể “trợ giúp” cho tế bào B và kích thích sản xuất kháng thể như đã nêu ở trên, cũng như thúc đẩy phản ứng của tế bào T CD8 tác động và kích thích đại thực bào cư trú trên đảo nhỏ [7].
Tài liệu tham khảo cho dịch bài
1.Vinmec (2019) Cơ chế tự miễn dịch trong bệnh tiểu đường loại 1,
https://www.vinmec.com/vi/tin-tuc/thong-tin-suc-khoe/co-che-tu-mien-dich-trong-benh-tieu-duong-loai-1/?link_type=related_posts , Updated September 02, 2020.
2. Ji-Won Yoon* and Hee-Sook Jun (2005) Autoimmune Destruction of Pancreatic b Cells, American Journal of Therapeutics 12, 580–591 (2005), page 580. Available in http://www.columbia.edu/itc/hs/medical/pathophys/immunology/readings/IsetCellDestructionReview.pdf, updated September 02, 2020.
3. Gabriel Espinosa-Carrasco, Cécile Le Saout, Pierre Fontanaud, Thomas Stratmann, Patrice Mollard, Marie Schaeffer and Javier Hernandez1 (2018) CD4+ T Helper Cells Play a Key Role in Maintaining Diabetogenic CD8+ T Cell Function in the Pancreas, https://www.frontiersin.org/articles/10.3389/fimmu.2017.02001/full, updated September 02, 2020.
4. MICHAEL O. KURRER, SYAMASUNDAR V. PAKALA, HOLLY L. HANSON, AND JONATHAN D. KATZ (1997) β cell apoptosis in T cell-mediated autoimmune diabetes, Proc. Natl. Acad. Sci. USA, Vol. 94, pp. 213–218, January 1997, Immunology. Xem tại: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC19288/ , Updated September 02, 2020.
5. Ramiro Diz., Alaina Garland., Benjamin G. Vincent, Mark C. Johnson, Nicholas Spidale, Bo Wang, Roland Tisch (2012) Autoreactive Effector/Memory CD4+ and CD8+ T Cells Infiltrating Grafted and Endogenous Islets in Diabetic NOD Mice Exhibit Similar T Cell Receptor Usage, Department of Microbiology and Immunology, University of North Carolina, Chapel Hill, North Carolina, United States of America, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0052054 , updated September 02, 2020.
6. Dieter Rondas, Valborg Gudmundsdottir, Wannes D'Hertog, Inne Crèvecoeur, Etienne Waelkens, Soren Brunak, Chantal Mathieu, Lut Overbergh (2015) A proteomic study of the regulatory role for STAT-1 in cytokine-induced beta-cell death, https://pubmed.ncbi.nlm.nih.gov/25712914/ , Updated September, 04 2020.
7. Adam L. Burrack, Tijana Martinov and Brian T. Fife (2017) T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5723426/ , Updated September, 04 2020.
Doctor SAMAN
Vũ Khắc Lương
Phó giáo sư, Tiến sĩ Y học, Giảng viên cao cấp Trường Đại học Y Hà Nội





















.png)




.png)
.jpg)






































.jpg)




